Estudiarem molts possibles nous fàrmacs similars o derivats de fàrmacs coneguts emprant diferents eines: QSAR, docking molecular i recerca amb algorismes aprenentatge profund amb diverses biblioteques python per a la recerca in silico de nous fàrmacs que actuen sobre diverses proteïnes disponibles al Protein Data Bank.
Pots dibuixar amb Avogadro i/o altres editor químics online (online chemical editors). Des de Avogadro, Afegir Hidrògens i Optimitzar geometria o estructura i guardar com mol2. Tambe pots transformar compostos químics des de Pubchem (smiles, sdf), Chemspider o Base de dades de productes naturals Coconut(mol), Zinc database (sdf) mitjançant Avogadro o amb Conversor formats químics
Base de dades de productes naturals de Medicina Tradicional Xinesa de Taiwan
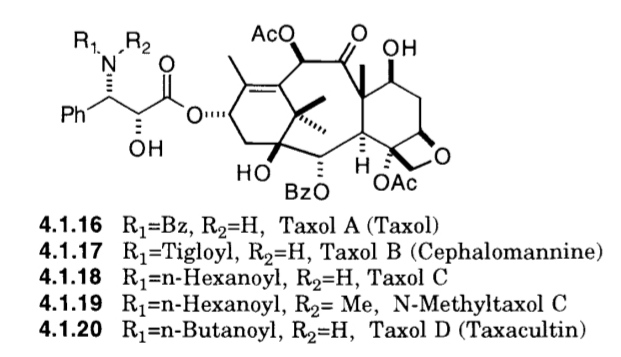
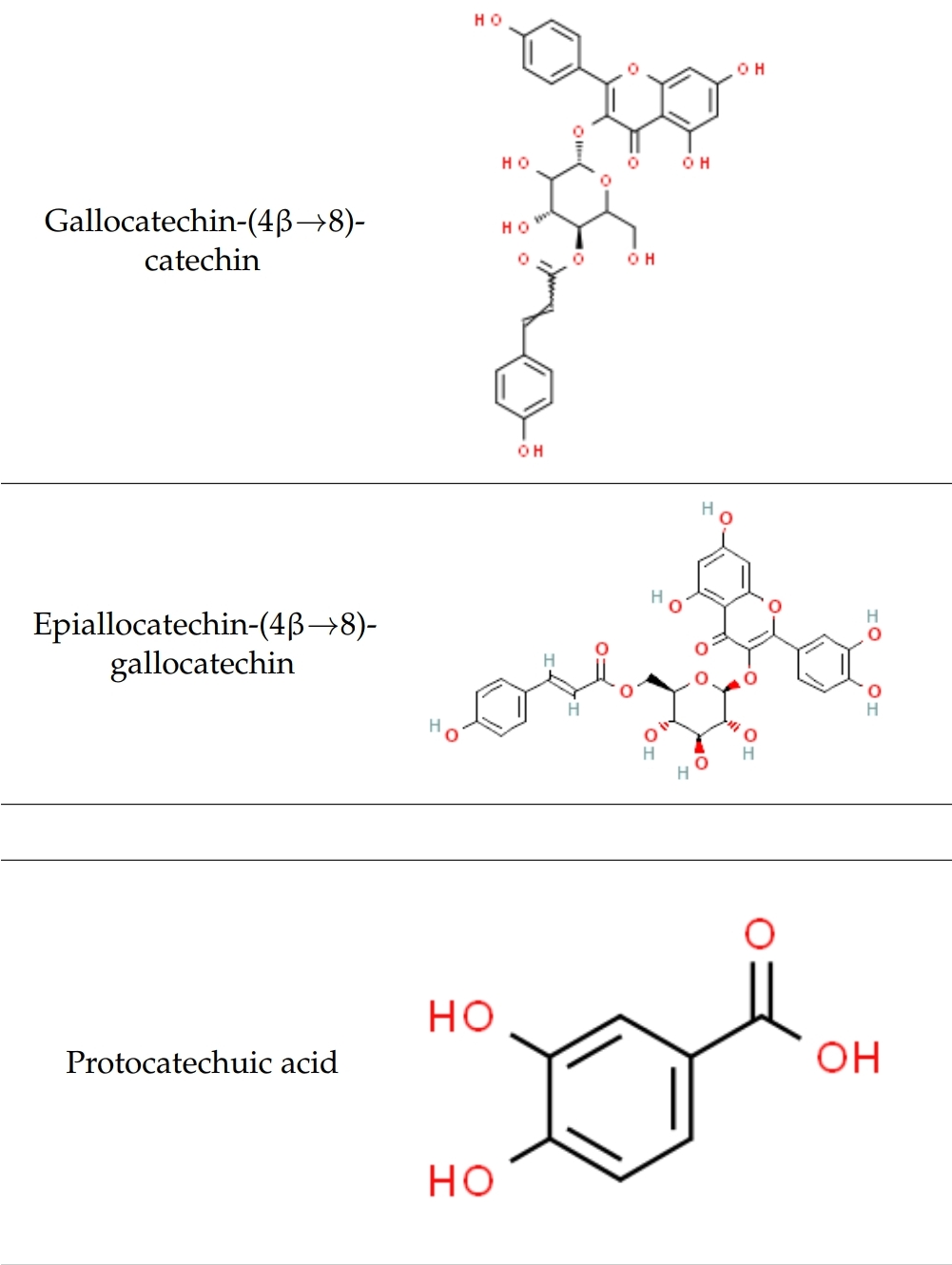
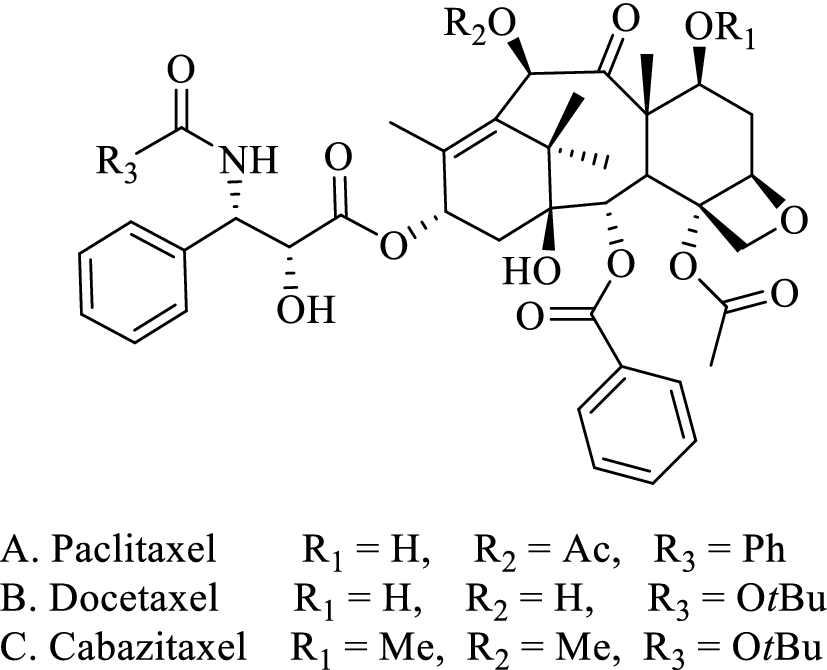
Taxans anticancerígens aprovats: paclitaxel (taxol), docetaxel, cabazitaxel
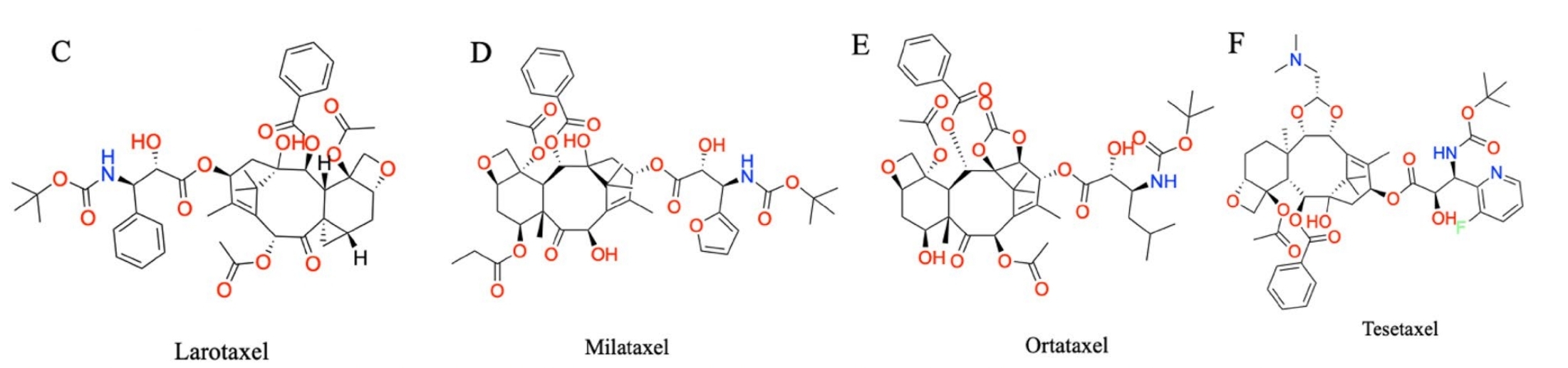
Altres taxans en estudis clínics en curs: larotaxel, milataxel, ortataxel i tesetaxel
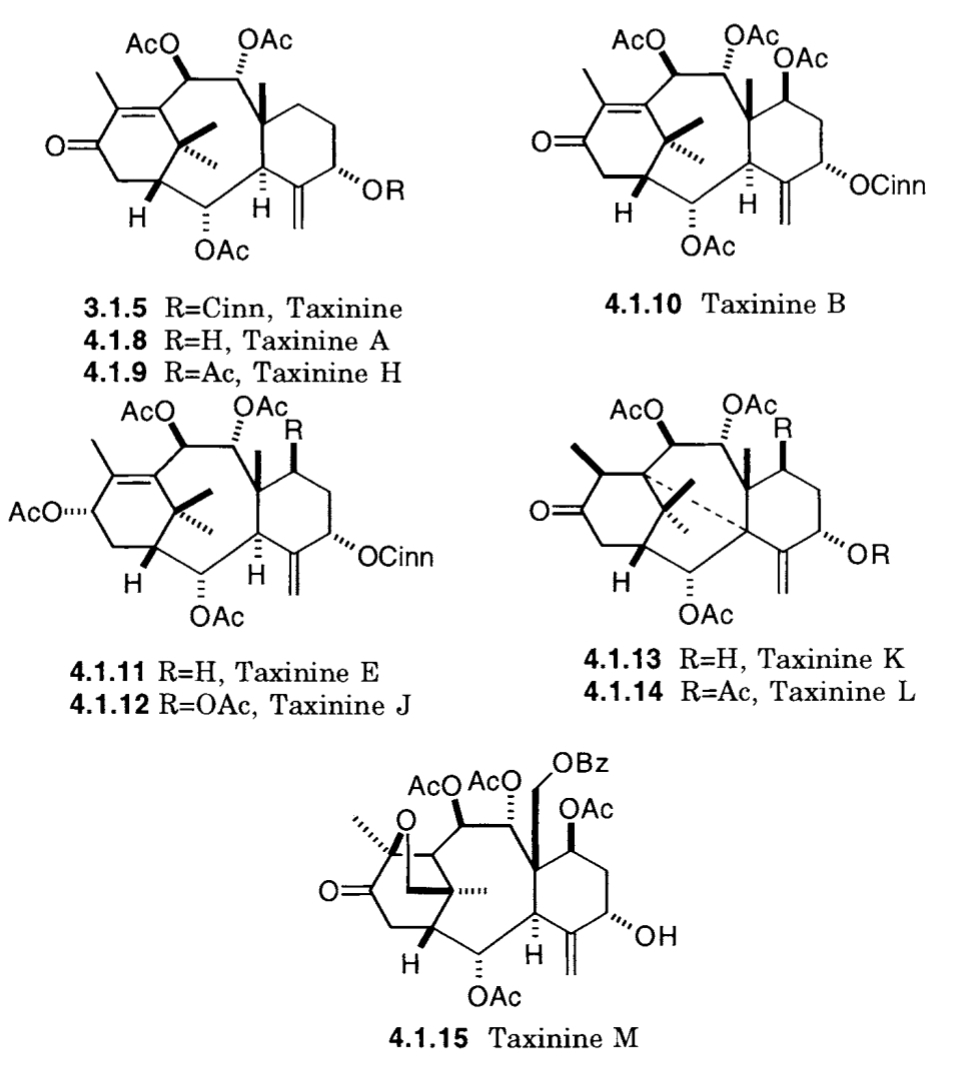
Altres taxans com les taxinines: taxinine A, B, C, J, H, K, etc.
Altres taxans com la família de les taxines: taxine A, B i C
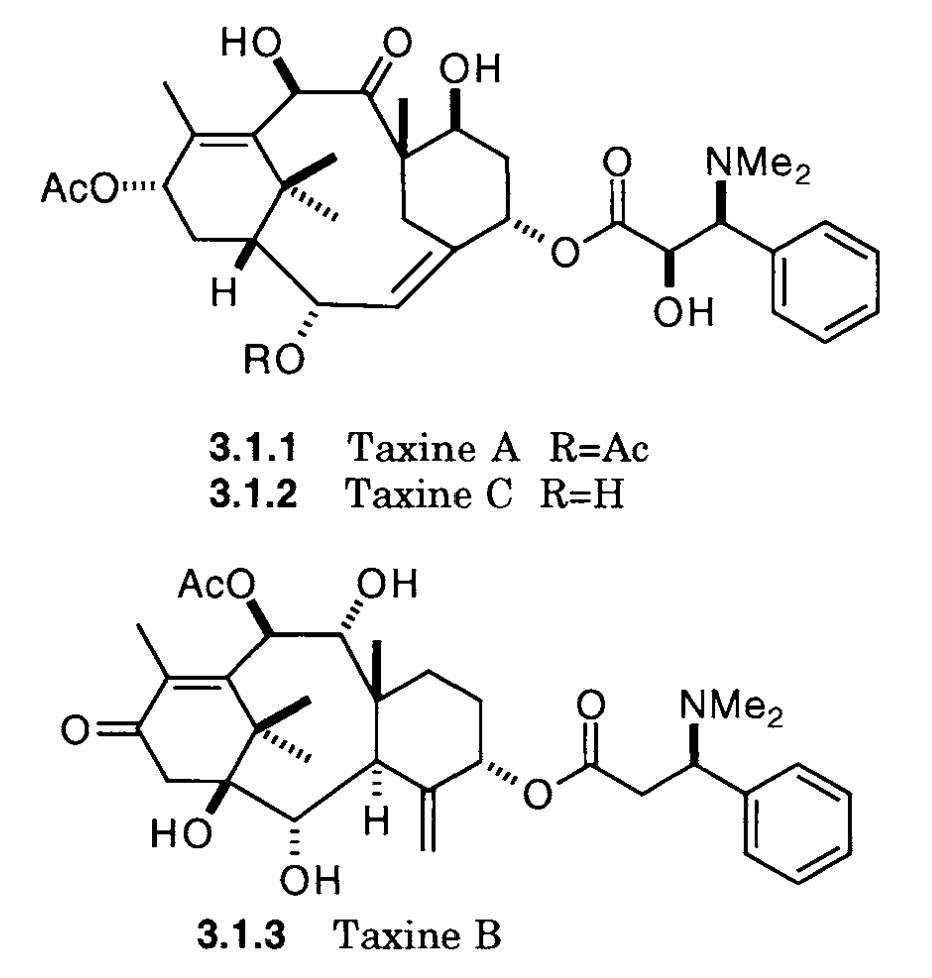
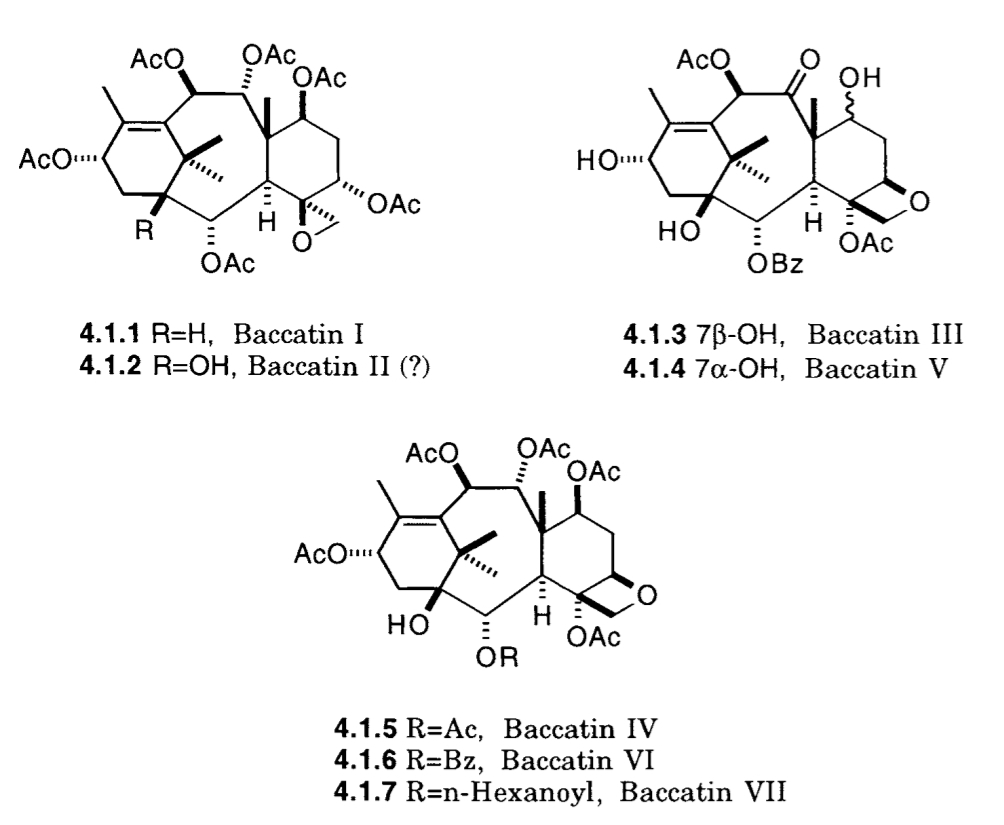
I, de moment, la darrera família: les baccatines
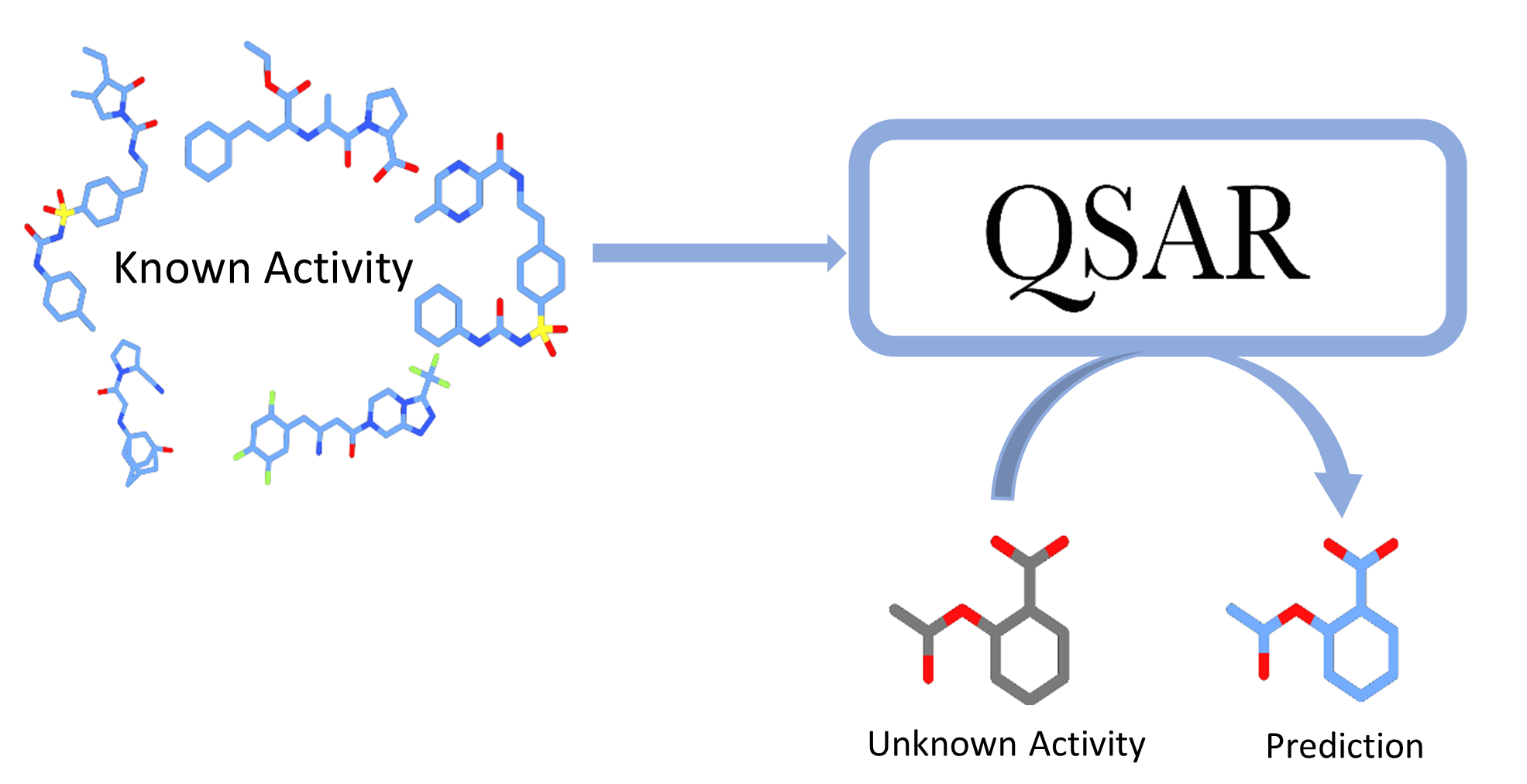
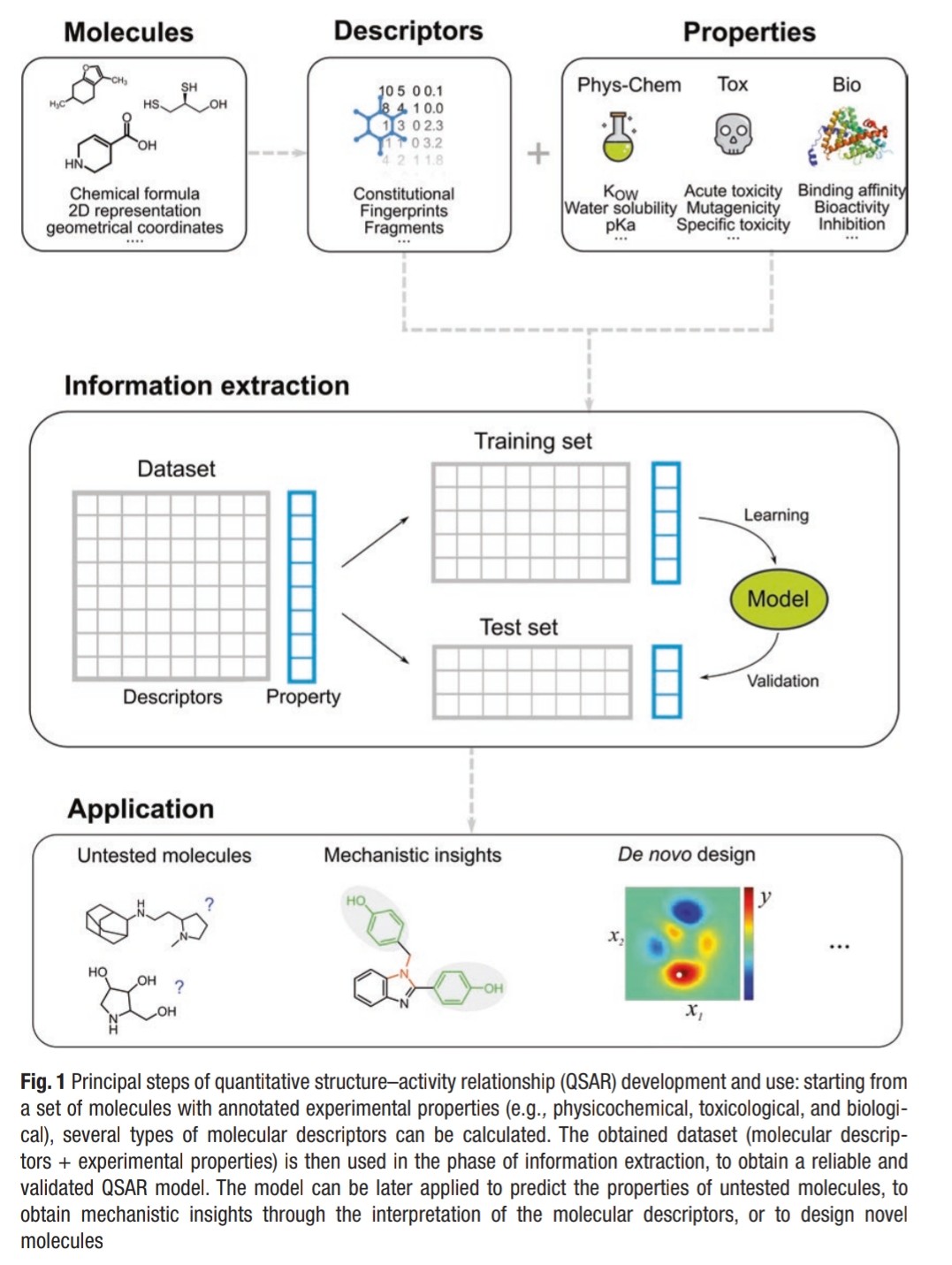
QSAR (Quantitative Structure-Activity Relationship) és un mètode utilitzat en la química computacional per predir i comprendre la relació entre la estructura química d'una substància i la seva activitat biològica o propietats físico-químiques. Aquesta tècnica implica l'ús de models estadístics o computacionals per correlacionar les característiques moleculars amb l'activitat observada experimentalment. QSAR és útil en el desenvolupament de fàrmacs, la toxicologia, el disseny de productes químics i altres aplicacions relacionades amb la química i la biologia.
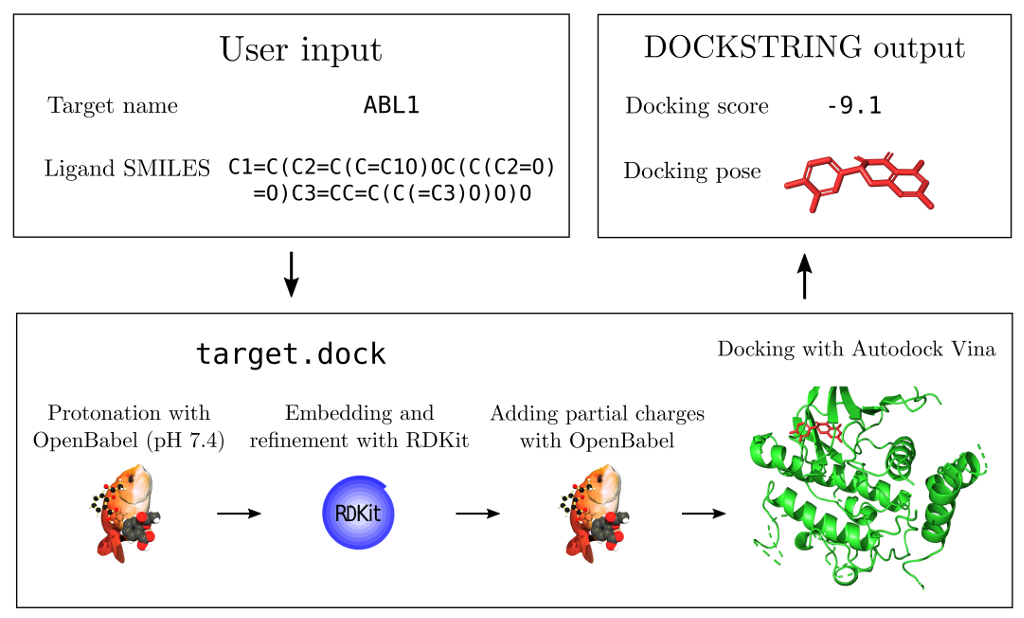
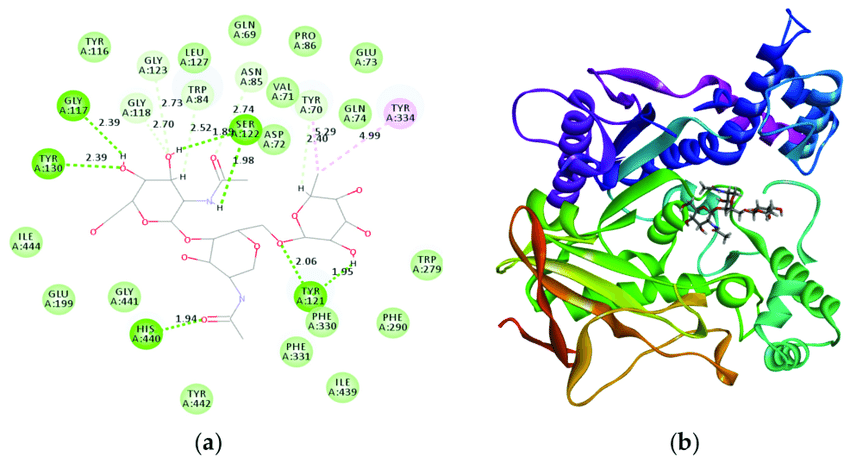
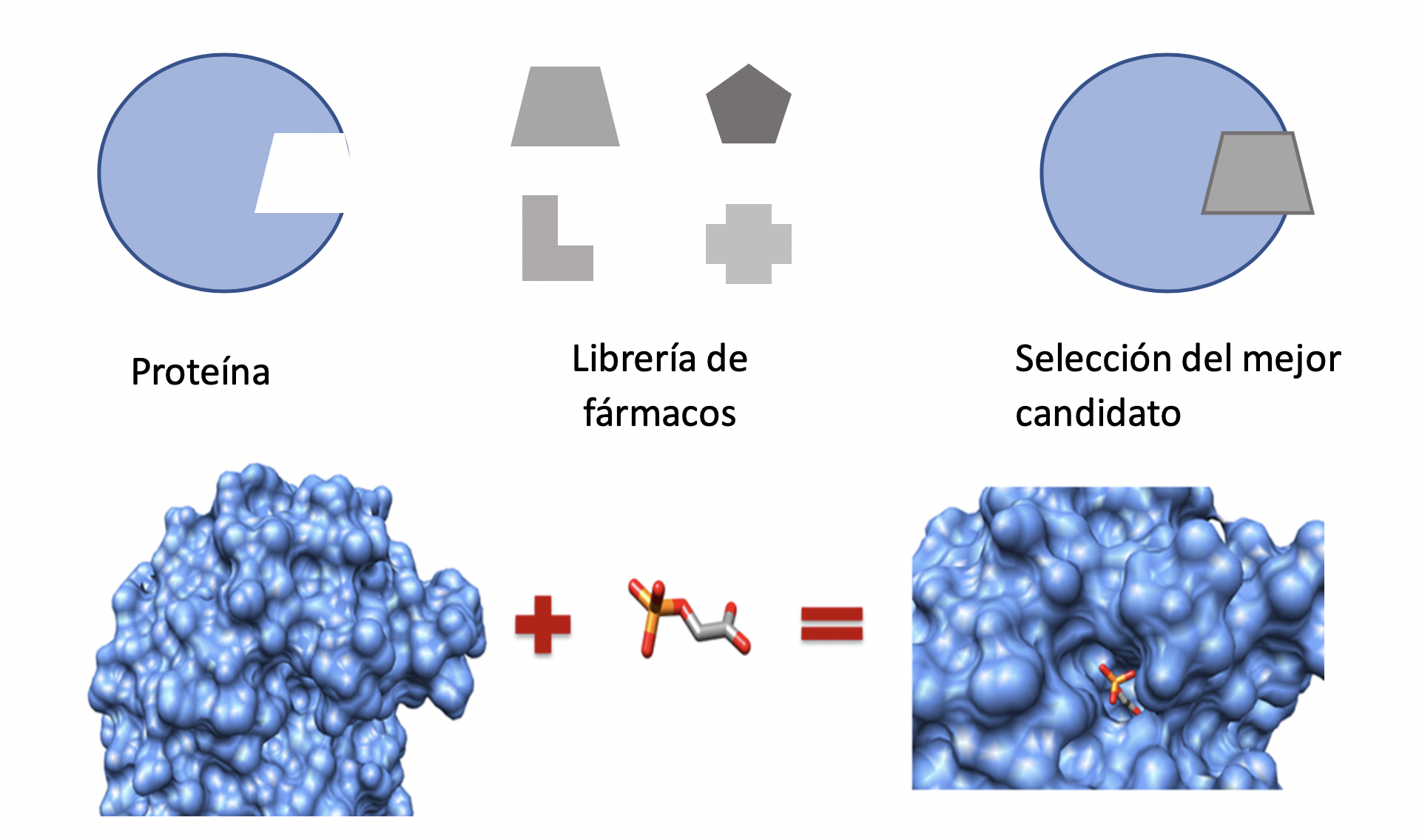
El docking molecular és una tècnica computacional utilitzada en química i biologia per predir com interactuen dues molècules, com ara un possible fàrmac i una proteïna, a nivell molecular.
Els descriptors moleculars són característiques quantitatives que es calculen per descriure una molècula emprades en QSAR. Aquí tens els diferents tipus:
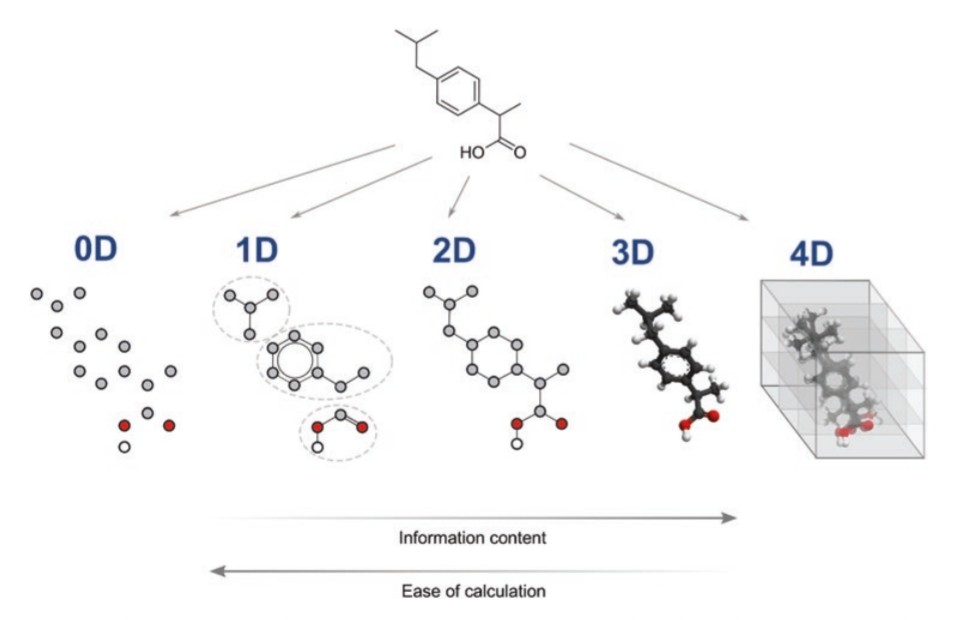
Els descriptors 0D són valors únics que descriuen les propietats globals d'una molècula, com ara la massa molecular o el nombre d'àtoms.
Els descriptors 1D són valors calculats a partir de l'estructura lineal d'una molècula, com ara les empremtes digitals que indiquen amb matrius de 0 i 1 la presència o absència de caracteristiques, subestructures o grups funcionals.
Els descriptors 2D tenen en compte l'estructura plana de la molècula, com ara la topologia de l'estructura molecular o els índexs de Wiener. Els descriptors 2D típics són la matriu d'adjacència, la matriu de Coulomb o la matriu de distància. En el cas de la matriu d'adjacència, el descriptor indica quins àtoms estan units en una molècula. Com que els descriptors 2D són sensibles a les característiques estructurals de la molècula (mida, forma i simetria), són una opció habitual com a descriptors moleculars.
Els descriptors 3D tenen en compte la forma tridimensional de la molècula, com ara el volum o l'àrea de superfície de la molècula. Els descriptors 3D més coneguts són la matriu molecular i els descriptors 3D-MoRSE. En el cas de la matriu molecular, el descriptor representa les coordenades cartesianes (x, y, z) de cada àtom. Aquests descriptors proporcionen molta informació sobre les molècules i tenen l'avantatge de diferenciar molècules isomèriques, cosa que no és el cas de tots els descriptors. Tanmateix, a causa de la seva complexitat, el càlcul dels descriptors geomètrics pot requerir molt de temps.
Els descriptors 4D són descriptors dinàmics que poden canviar amb el temps o en resposta a factors externs, com ara descriptors de flexibilitat molecular o descriptors de reactivitat. Els descriptors 4D també s'anomenen "descriptors basats en quadrícula". Aquests descriptors, a més de la geometria molecular, introdueixen una quarta dimensió. Aquesta nova dimensió normalment caracteritza les interaccions entre la o les molècules i el lloc o els llocs actius d'un receptor o els múltiples estats conformacionals de la o les molècules. Els descriptors 4D comuns són CoMFA i GRID. Un avantatge dels descriptors 4D és que proporcionen més informació que els altres descriptors i sempre són capaços de generar valors diferents per a molècules estructuralment diferents. Tanmateix, com els descriptors 3D, els descriptors 4D no són fàcils d'obtenir a causa de la seva major complexitat.
 Descriptors moleculars i empremtes digitals amb Padelpy
Descriptors moleculars i empremtes digitals amb Padelpy
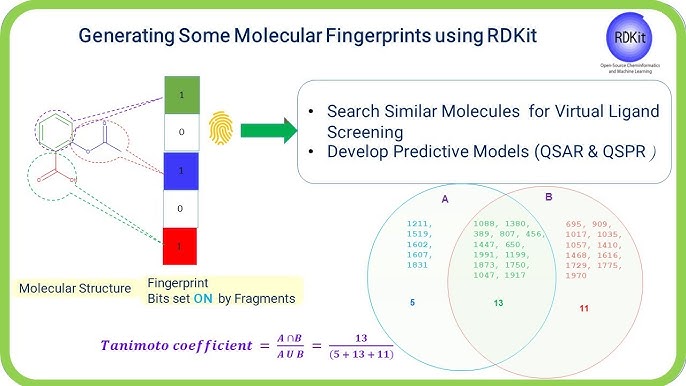
Amb milers de descriptors moleculars existents, seleccionar els descriptors més adequats és pot fer mitjançant la cerca exhaustiva i els algorismes d'optimització. La cerca exhaustiva, també coneguda com a Model de Subconjunts Totals (ASM) consisteix en la generació de totes les combinacions possibles per a N descriptors a provar, es proven 2^N-1 combinacions de descriptors. Altres estratègies són els algorismes d'optimització, procediments iteratius que es poden utilitzar per trobar la combinació òptima de descriptors d'un conjunt de descriptors moleculars (que condueix al millor model predictiu per a les propietats estudiades). Els algorismes d'optimització més comuns utilitzats en la selecció dels descriptors són la programació evolutiva, l'optimització de colònies de formigues, la cerca seqüencial i els algorismes genètics.
En el cas del docking molecular hem de tenir en compte:
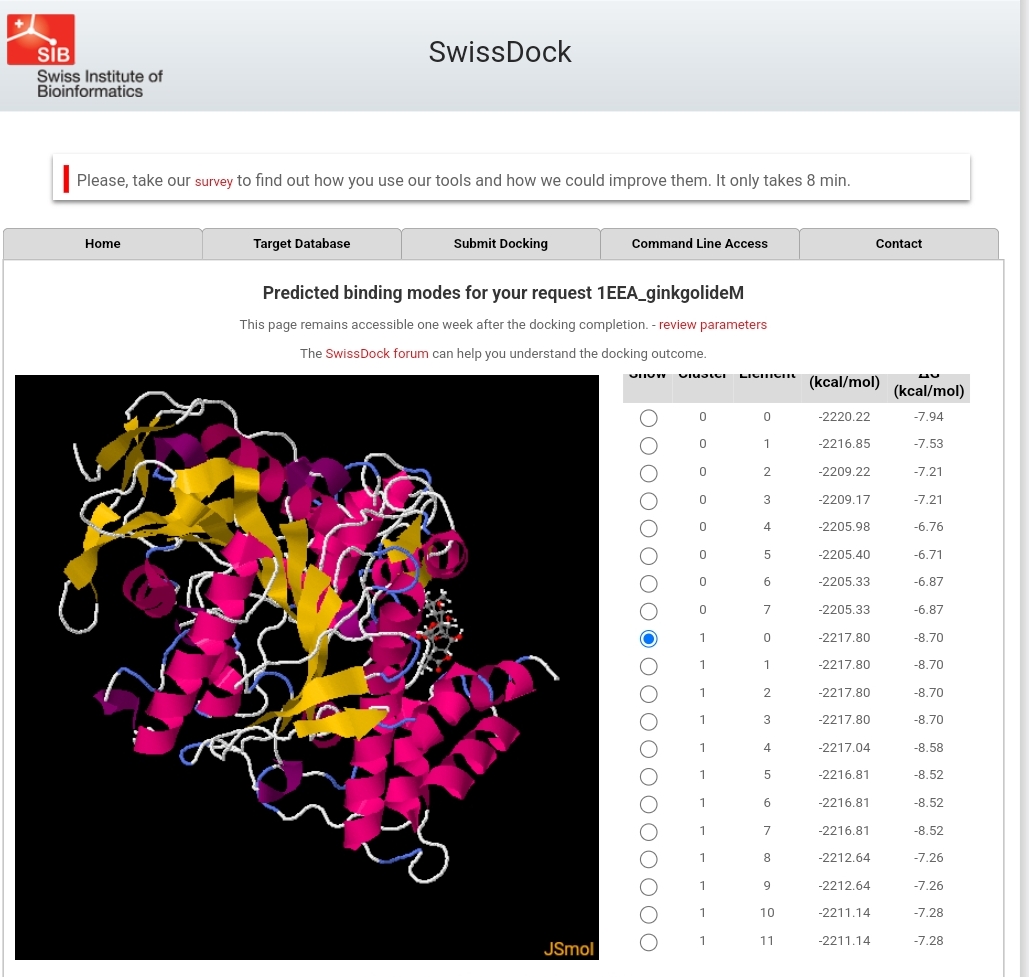
Per buscar i preparar un fàrmac en un format adequat per a SwissDock, segueix aquests passos:
Per preparar un fàrmac amb Avogadro, segueix aquests passos:
Per convertir fitxers SDF i SMILES a MOL2:
Assegura't que el fitxer MOL2 resultant compleixi amb els requisits de SwissDock, incloent la presència de tots els hidrogens i càrregues correctes.
Un cop preparats, segueix aquests passos per a la submissió a SwissDock:
Per a més informació, consulta la Guia d'Usuari de UCSF ChimeraX i la pàgina de SwissDock.
Utilitza el meu Google colab per analitzar les dades i obtenir gràfics com aquest
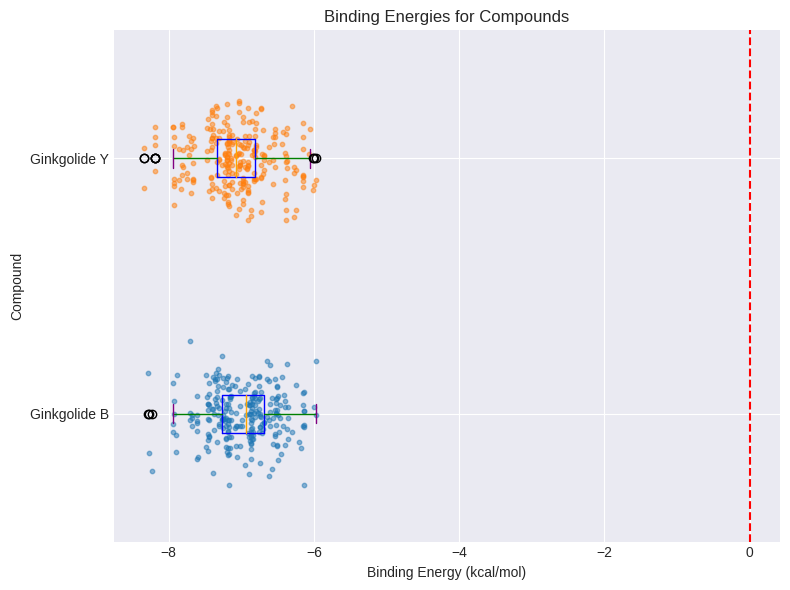
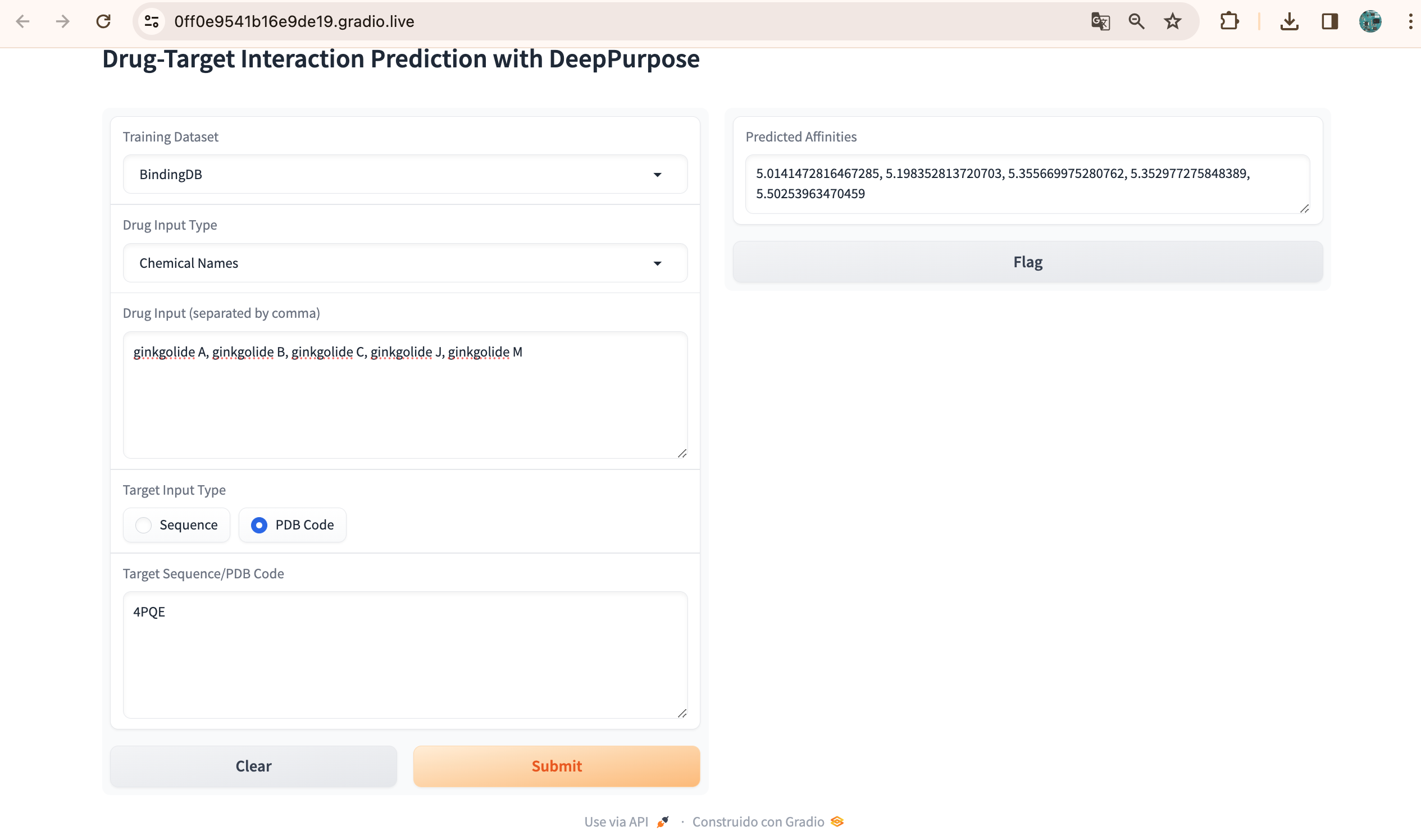
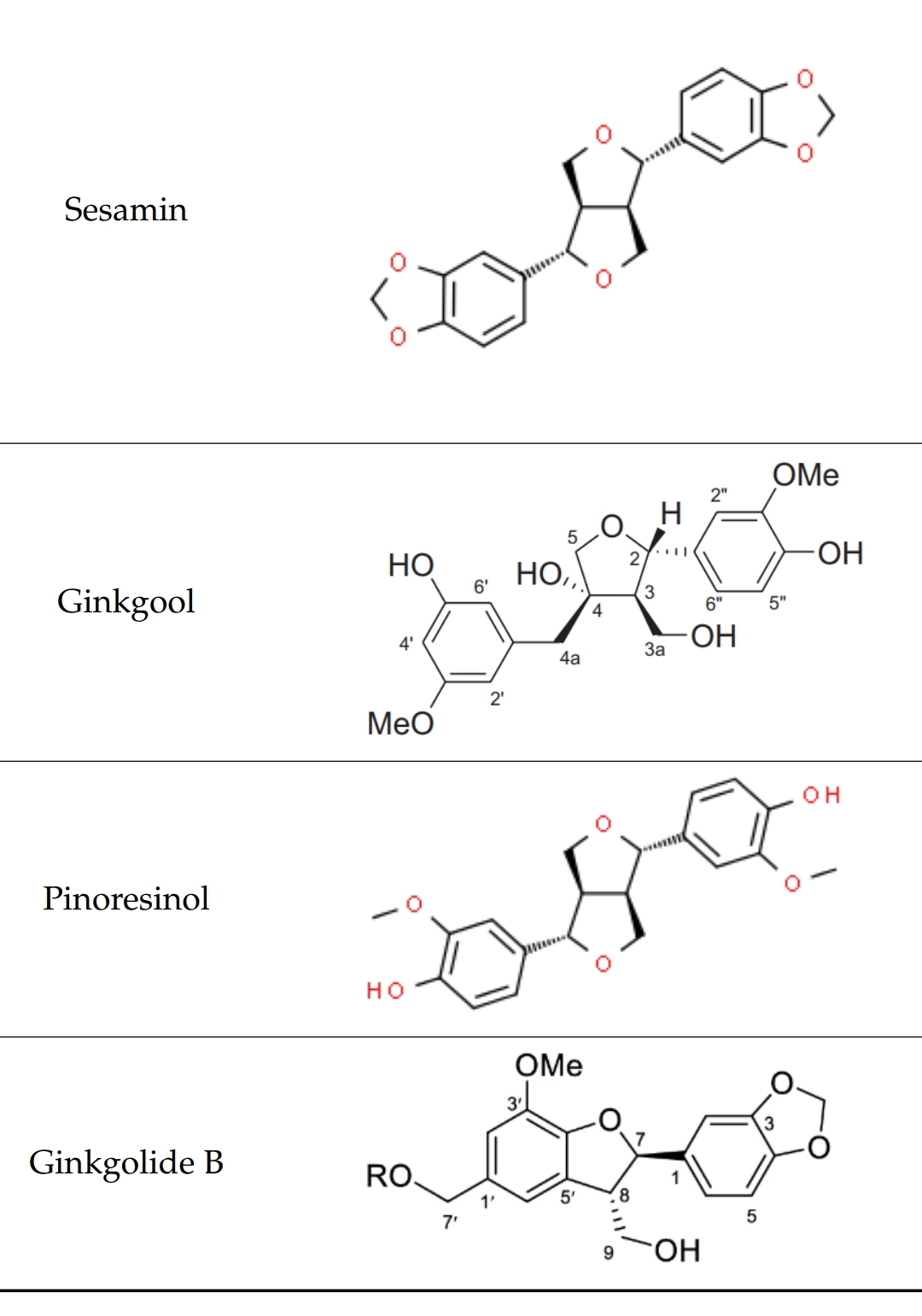
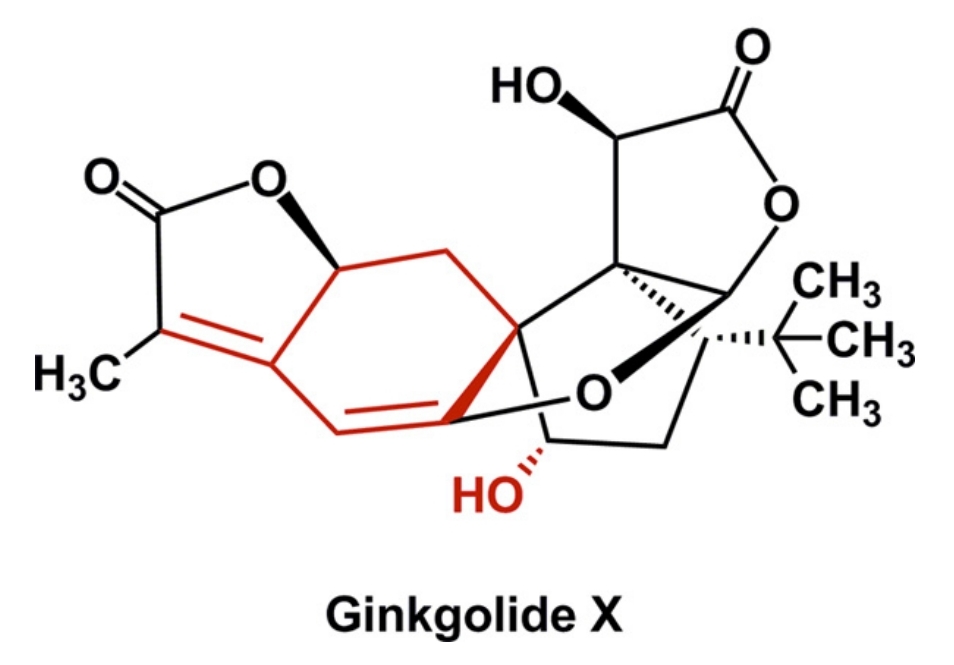
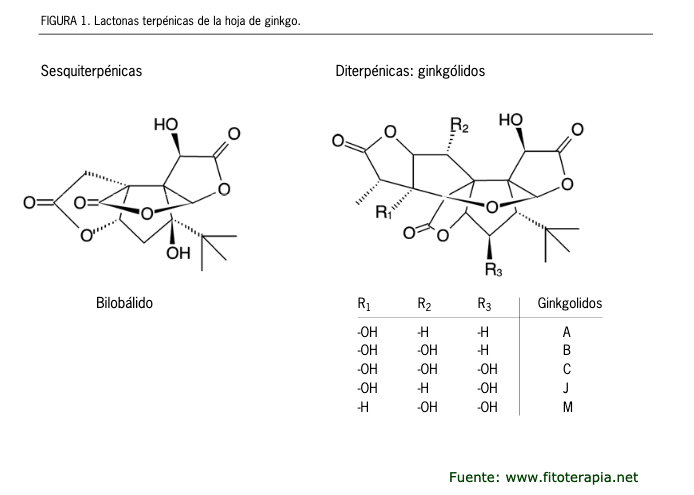
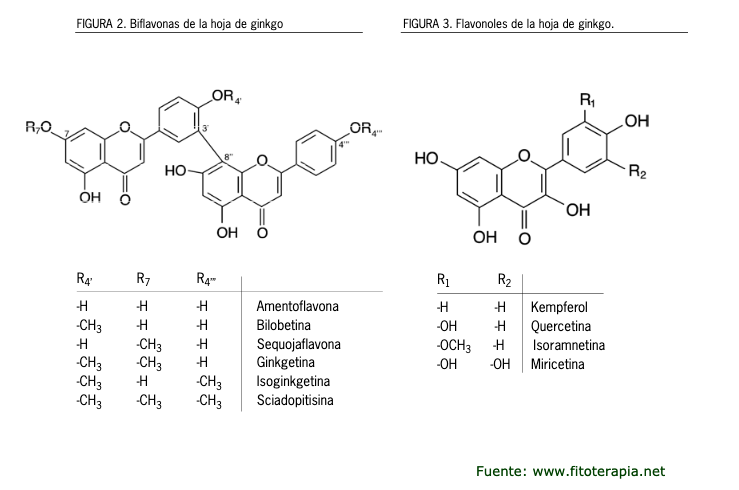
En la recerca de nous fàrmacs, es realitza el docking molecular per predir com interaccionen compostos com els ginkgòlids amb la enzima acetilcolinesterasa, que està implicada en diversos processos biològics com per exemple els trastorns cognitius.
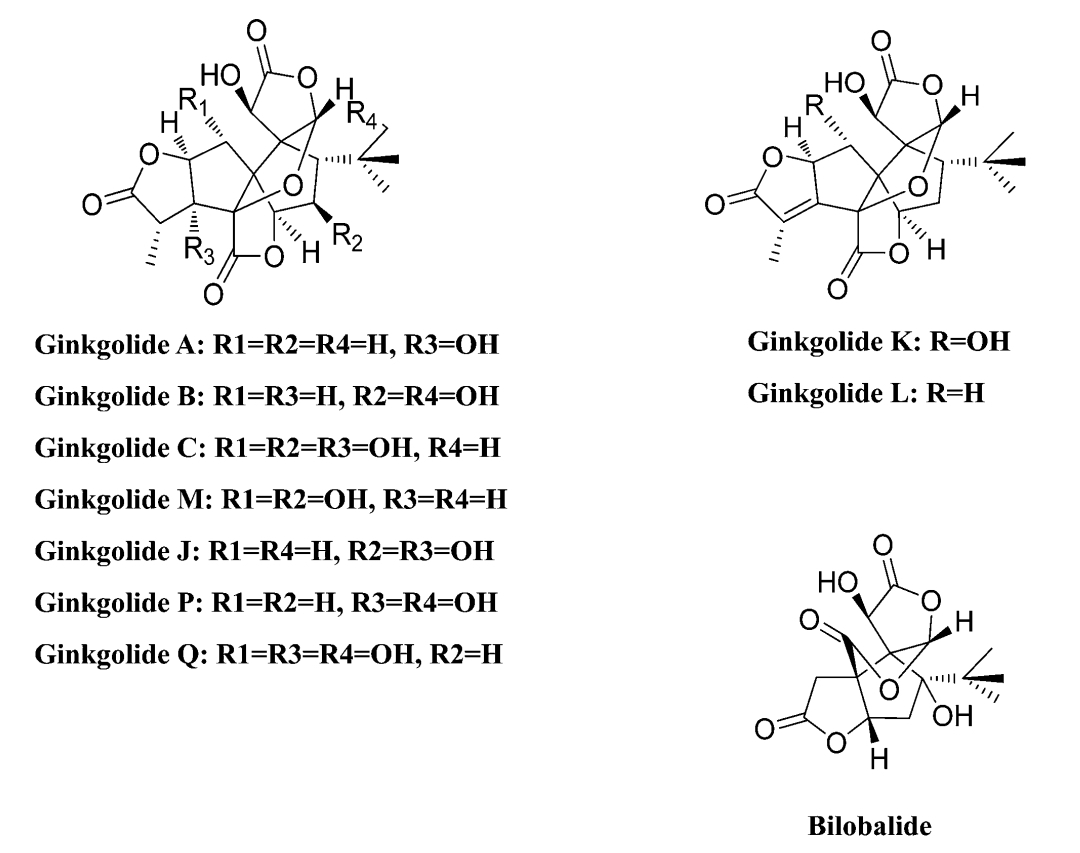
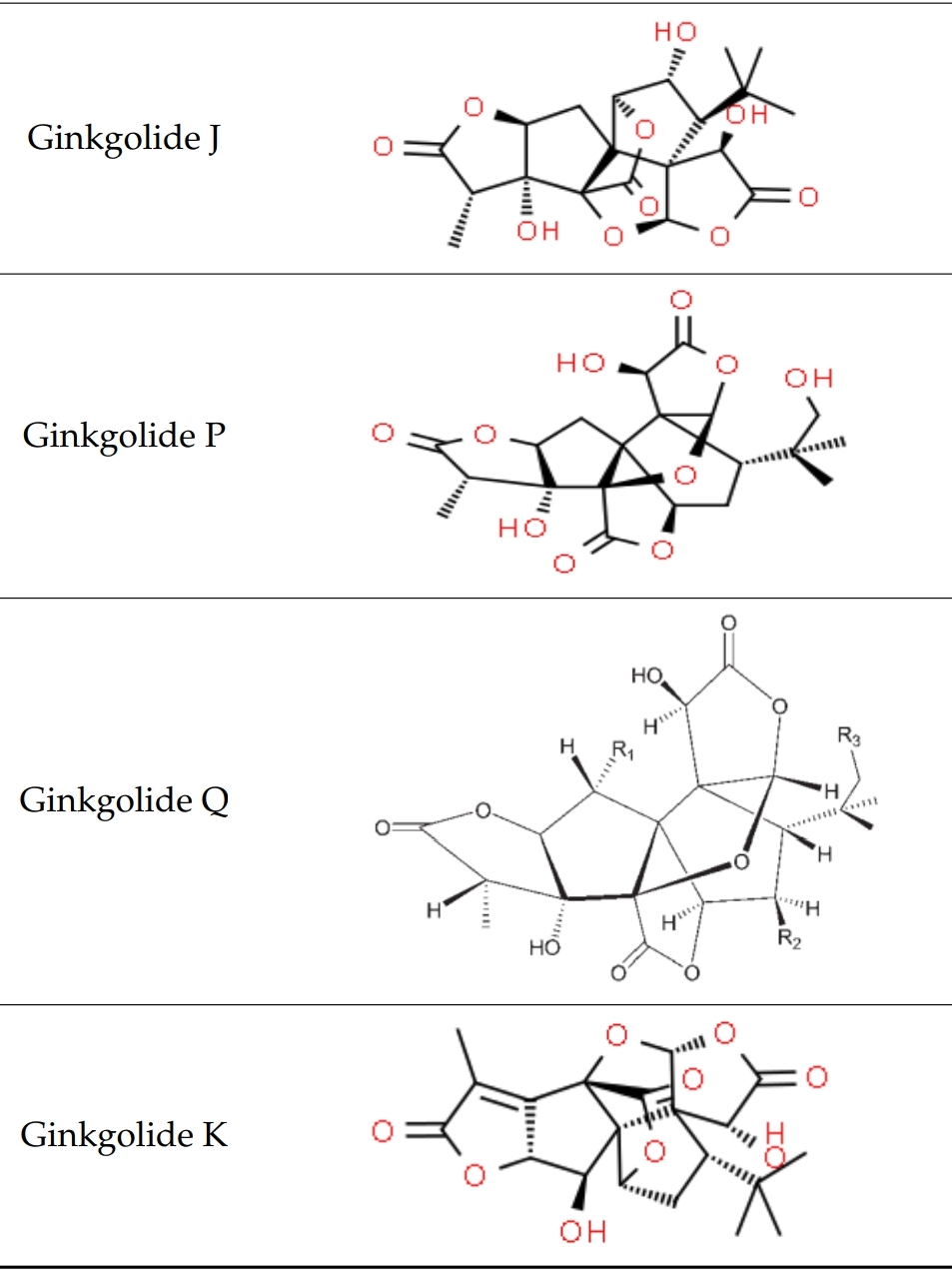
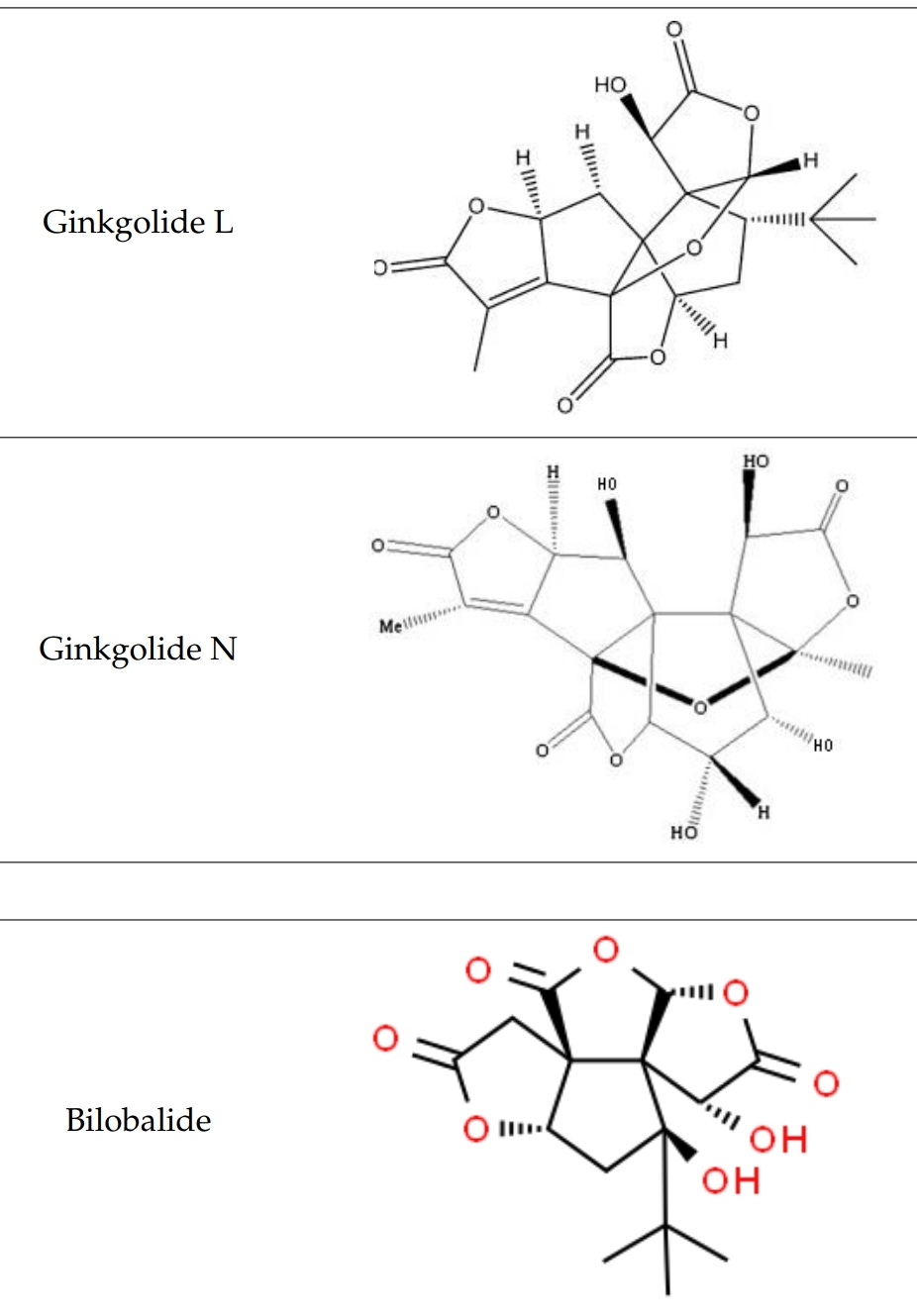
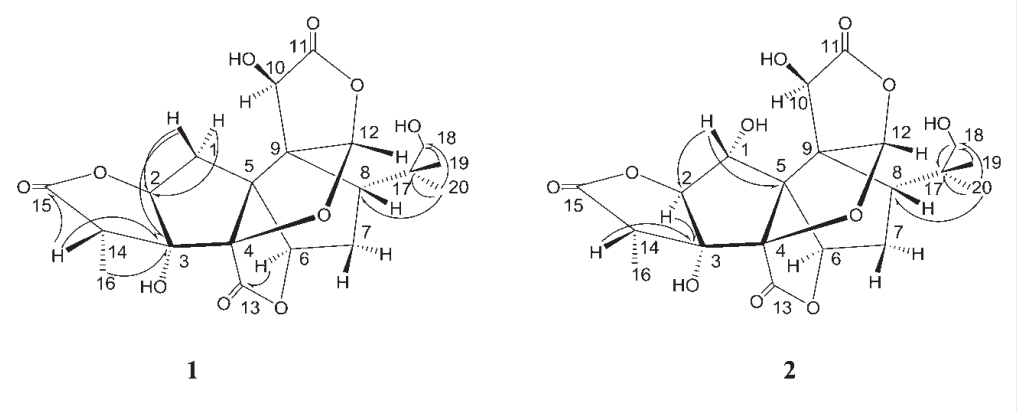
Ginkgolide P(1) i Ginkgolide Q(2)
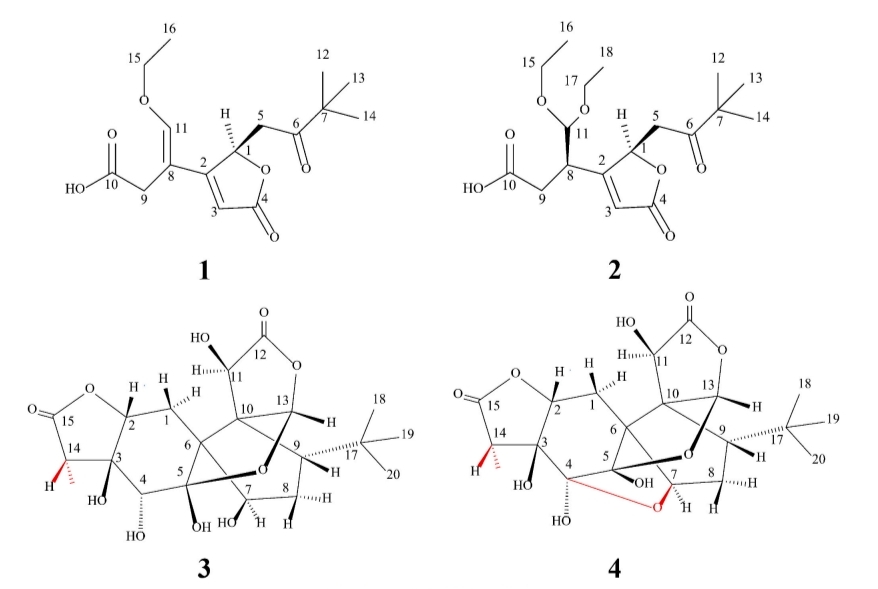
Bilobalide A (1), bilobalide B (2), ginkgolide W (3), and ginkgolide Y (4)
Els taxans són una classe de compostos utilitzats en quimioteràpia per combatre el càncer. El docking molecular es pot utilitzar per comprendre com aquests taxans interactuen amb la tubulina, una proteïna implicada en la divisió cel·lular.
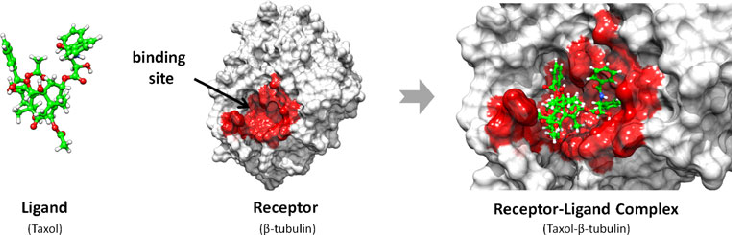
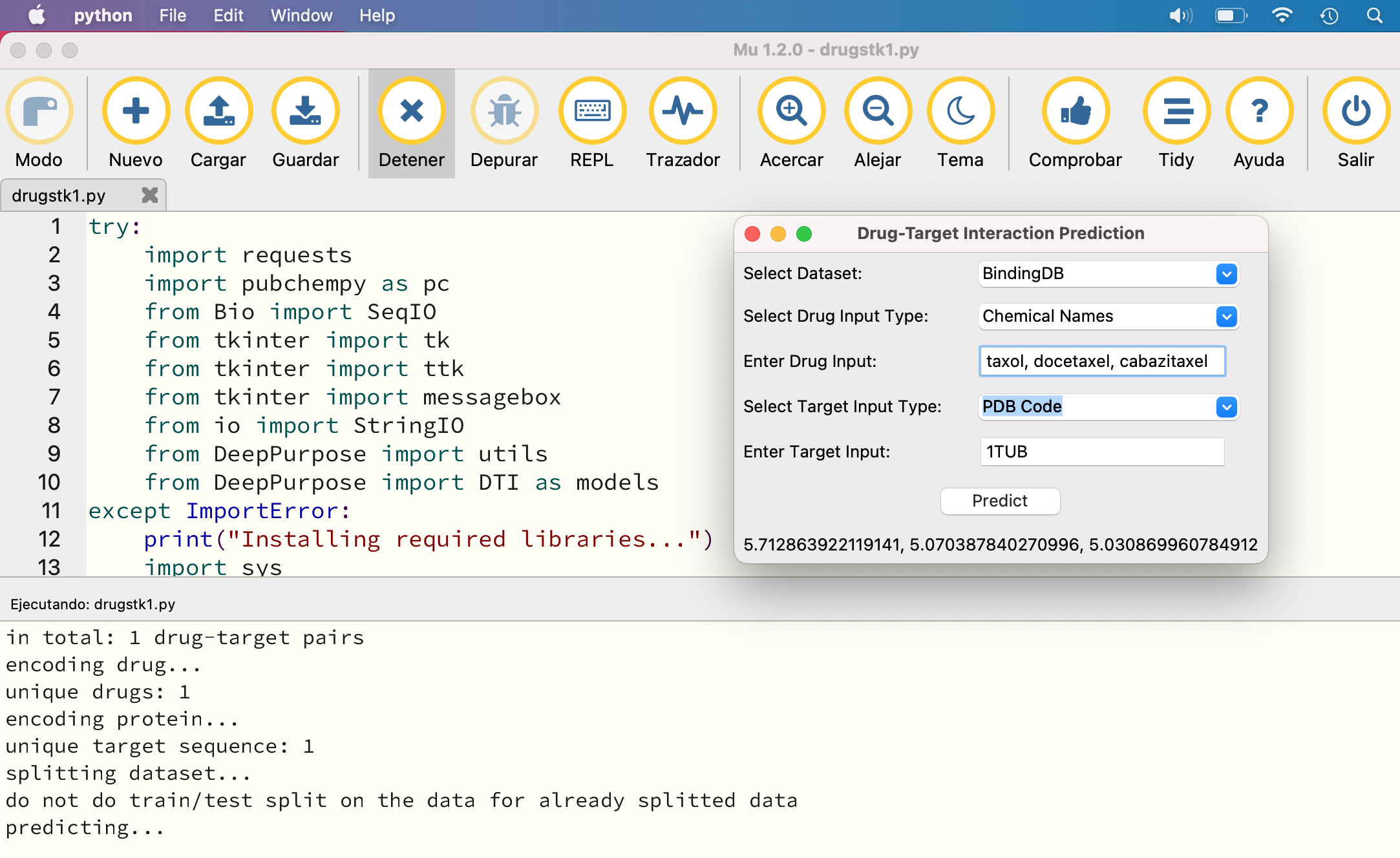
El projecte que utilitza DeepPurpose i Dockstring en Python es relaciona amb el desenvolupament de noves eines computacionals per a la recerca en química i biologia molecular. DeepPurpose és una llibreria de Python que ofereix models d'aprenentatge profund pre-entrenats per a la predicció de l'activitat biològica de molècules, com ara la interacció amb proteïnes diana, com la acetilcolinesterasa en el cas dels ginkgolids o toxines russes. D'altra banda, Dockstring és una eina que facilita el docking molecular, és a dir, la predicció de la forma i l'afinitat de les molècules químiques quan interaccionen amb les proteïnes. En resum, aquest projecte integra les capacitats predictives de DeepPurpose amb les funcionalitats de Dockstring per a realitzar anàlisis molecular avançats i accelerar la descoberta de nous compostos amb aplicacions en medicina i biotecnologia.
Per obtenir més informació sobre el projecte, podeu consultar la pàgina oficial de DeepPurpose per conèixer les capacitats dels seus models d'aprenentatge profund. A més, podeu accedir a la documentació de Dockstring per aprendre com utilitzar aquesta eina per al docking molecular.